发文章还差一口气?网络药理学+分子对接+分子动力学模拟,这条证据链帮你补上
2026-06-08
引 言
如果你关注过中药研究、天然产物发现或药物筛选领域,大概率对三个词不陌生:网络药理学、分子对接、分子动力学模拟。
它们频繁出现在近几年的论文中,有时单独出现,有时两两组合,有时三者串联。
但很多人对它们的理解是模糊的:分别解决什么问题?之间什么关系?一篇完整的研究应该怎样串联三者?
今天这篇推文,一次性讲清楚。
一、网络药理学:从“整体”出发的系统筛选
网络药理学是从系统层面研究药物与疾病之间关系的方法。
它的核心思路是:药物不是只作用于一个靶点,而是通过多个成分、多个靶点、多条通路协同发挥作用。
基于这一思路,网络药理学构建出“药物-成分-靶点-疾病-通路”的复杂网络,帮助我们理解药物作用的整体图景。
它回答的是:“哪些成分可能作用于哪些靶点?”
在药物研发的早期阶段,面对成千上万的可能性和未知机制,网络药理学提供了一个高效的筛选工具。
它把研究范围从“大海捞针”缩小到“几个候选对象”,为后续验证指明方向。
优势很明显:整体视角、高通量筛选、适合复杂药物体系(尤其是中药复方)的作用机制研究。
局限也同样存在:网络药理学的结果本质上是预测和关联,给出的是“可能性”而非“确定性”。
它告诉你A和B之间有边相连,但无法证明在真实生理条件下A真的能结合B并产生功能效应。
这正是后续方法需要介入的地方。
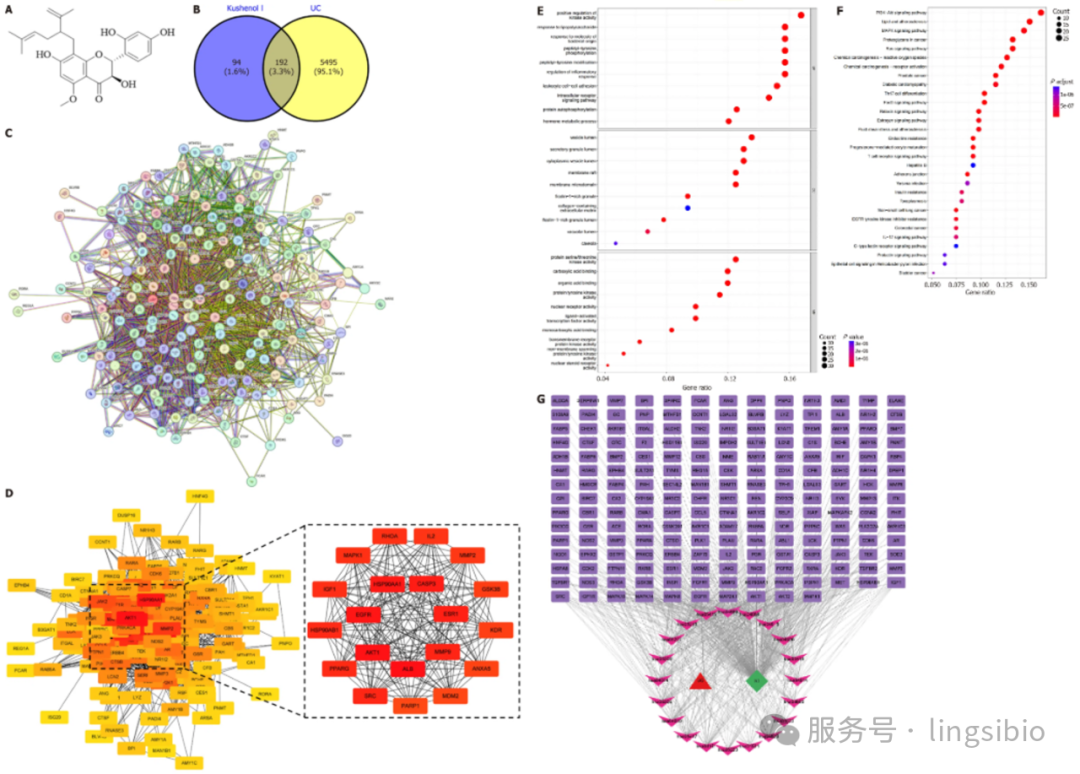
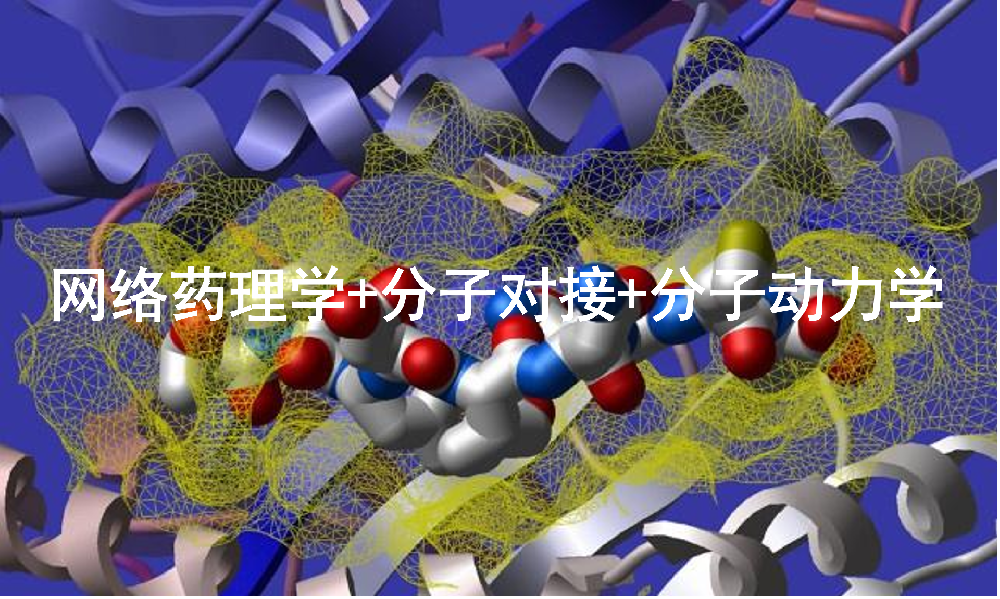
图1 网络药理学分析图
图例来源:
He X D, Li M, Zuo X D, et al. Kushenol I combats ulcerative colitis via intestinal barrier preservation and gut microbiota optimization[J]. World Journal of Gastroenterology, 2025, 31(26): 105656.
二、分子对接:从“结构”出发的结合验证
分子对接是一种基于三维结构的计算方法。它模拟药物分子(配体)与靶点蛋白(受体)在空间上的相互识别与结合过程,通过搜索配体在受体结合口袋中的可能位置和取向,找到能量最优的结合构象。
简单说:把两个分子放到一起,看它们能不能“对上”。
它回答的是:“两者能不能结合?以什么方式结合?”
分子对接给出两个核心输出:
结合能:数值越低(负得越多),表示结合亲和力越强
结合模式:氢键、疏水作用、π-π堆积等相互作用的具体位置
这些信息在药物筛选中非常有价值:
结合能帮你排序候选分子,结合模式告诉你哪些结构特征是关键的,为后续结构优化提供直接依据。
优势是计算速度快、成本低、可批量处理。在已知靶点结构的情况下,分子对接是筛选化合物库的高效工具。
局限在于:分子对接本质上是静态的。
它把蛋白视为刚性或半柔性的结构,在相对有限的构象空间内搜索结合模式。
但真实生理环境中,蛋白和小分子都在持续热运动,结构是动态变化的。
分子对接给出的,更像是一张“瞬时快照”。
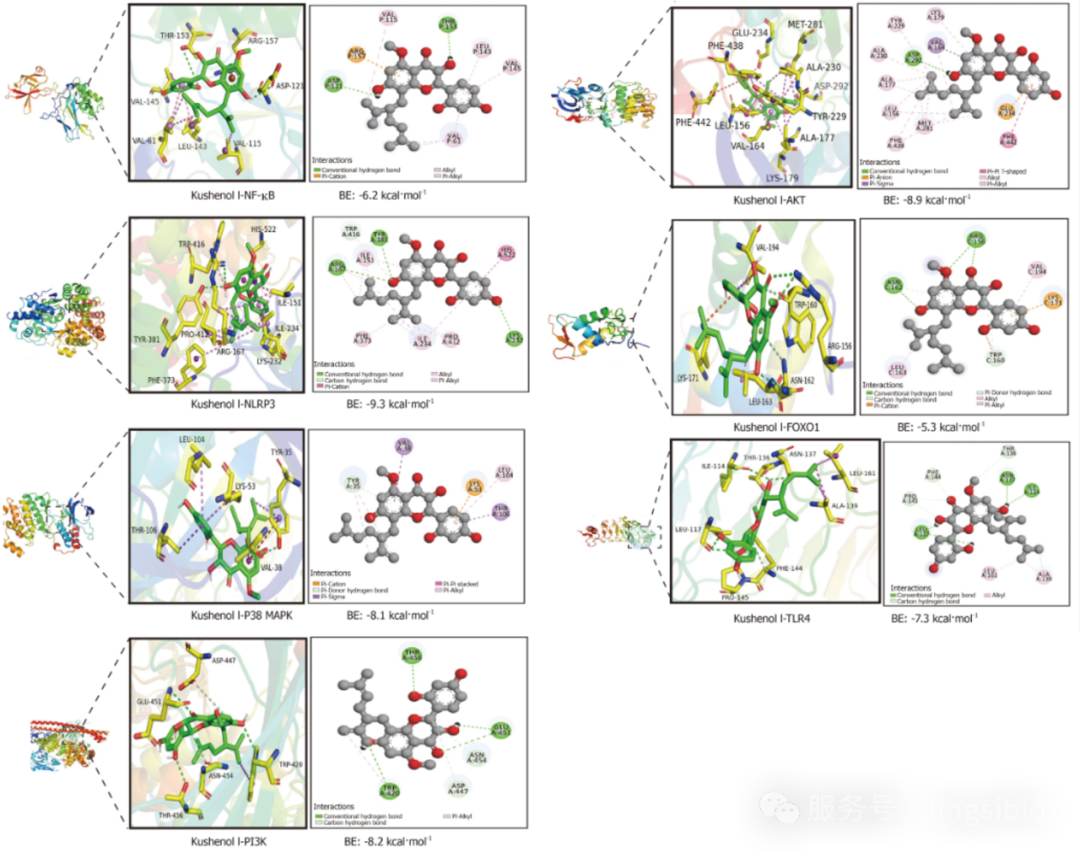
图2 分子对接模式图
图例来源:
He X D, Li M, Zuo X D, et al. Kushenol I combats ulcerative colitis via intestinal barrier preservation and gut microbiota optimization[J]. World Journal of Gastroenterology, 2025, 31(26): 105656.
三、分子动力学模拟:从“时间”出发的动态检验
分子动力学模拟是基于牛顿力学的计算方法。
它在原子尺度上模拟分子体系随时间演化的行为。研究者把复合物放入一个模拟的生理环境(水溶液、特定温度、特定压力)中,让计算机一步步计算每个原子的位置和速度变化,最终得到一条完整的运动轨迹。
可以理解为:给分子复合物录一段视频,看它在真实环境下的动态表现。
它回答的是:“结合之后稳不稳?能维持多久?”
分子动力学模拟提供的信息远比静态结构丰富:
RMSD:监测复合物整体结构的稳定性,看它是否在模拟过程中保持收敛
RMSF:识别蛋白上哪些残基在结合后变得“僵硬”或保持“灵活”
氢键分析:追踪关键相互作用的持续性,而非只看瞬时的形成与否
回转半径:判断结合后蛋白结构是否变得更加紧实
结合自由能计算:更精确地评估结合强度,并可拆解为范德华贡献、静电贡献、溶剂化贡献等分量。
优势在于引入了时间维度和生理环境。一个对接结果如果能在100纳秒甚至微秒级别的模拟中保持稳定,它的可信度远高于单纯的对接打分。
此外,动力学模拟还能揭示构象变化、识别别构位点、捕捉结合与解离的过程细节。
局限是计算成本高。模拟时长、体系大小、力场选择都会影响结果质量。短时间模拟(如10纳秒)可能还没达到平衡就已结束,结论缺乏说服力。
图3 分子动力学模拟结果
图例来源:
He X D, Li M, Zuo X D, et al. Kushenol I combats ulcerative colitis via intestinal barrier preservation and gut microbiota optimization[J]. World Journal of Gastroenterology, 2025, 31(26): 105656.
四、三者关系:谁也无法替代谁
理解了三个方法分别做什么,三者之间的关系就清楚了。
不是替代关系,是递进关系。

一个比喻可以帮助理解:
缺少第一步:你不知道该模拟哪个体系,大海捞针。
缺少第二步:没有结构层面的验证,结合与否只停留在推测。
缺少第三步:证据停留在静态快照,缺乏对动态稳定性的检验。
三者各司其职,谁也替代不了谁。
五、一篇完整研究的典型结构
当三者串联使用时,一篇论文的结构通常是这样的:
这种结构的好处是:证据链完整,从系统筛选到静态验证再到动态检验,层层递进,每一步都有明确的科学问题支撑。
图4 网络药理学-分子对接-分子动力学模拟联合分析技术路线
图例来源:
Zhang S, Shao Y, Jin R, Ma B. Combining Network Pharmacology, Molecular Docking, Molecular Dynamics Simulation, and Experimental Validation to Uncover the Efficacy and Mechanisms of Si-Miao-Yong-An Decoction in Diabetic Wound Healing. J Inflamm Res. 2025 Mar 19;18:4087-4101. doi: 10.2147/JIR.S506739.
六、常见误区与澄清
误区一:三者做其中一个就够了
事实是:单独任何一种都有局限。网药给不出结构证据,对接给不出动态证据,动力学无法独立完成筛选。三者结合才是完整的证据链条。
误区二:动力学会取代对接
事实是:不会。对接是动力学的必要前置步骤。没有对接给出的初始构象,动力学模拟无从开始。对接筛选出少数候选体系后,动力学才能进行深度验证。
误区三:三者都用上就是好文章
事实是:关键在于问题的匹配度和逻辑的严密性。是否使用三种方法,取决于研究问题本身。有些问题只需要对接就够了;有些问题确实需要三者串联。盲目堆砌方法反而画蛇添足。
写在最后
网络药理学、分子对接、分子动力学模拟,分别代表了三种不同的科学视角:系统的、结构的、时间的。
把它们串联起来,不是为了“凑方法”,而是为了让证据链条更加完整、结论更加可靠。
 技术咨询:
技术咨询: