灵思研发团队组学相关—基因组
2022-11-17
武汉灵思生物
武汉灵思生物技术有限公司(Wuhan Lingsi Biotech Co.,Ltd.)成立于2015年11月,是一家专注于生命科学和生物技术领域的高科技企业。公司主要以细胞及实验动物为主要研究对象,利用常规分子生物学、细胞生物学及免疫学等技术联合高通量组学(基因组、蛋白组及代谢组)分析手段,推动疾病发病机制及药物疗效、药物作用靶点筛选及作用机制探讨。公司本着“设计合理、理念新颖、研发严谨、数据准确”的研究理念和不断创新、开拓进取的创业精神、通过高度细分、全方位的服务平台;帮助广大科研工作者在各研究领域突破贡献绵力,为生命科学的可持续发展提供强有力的帮助。
动植物基因组denovo测序
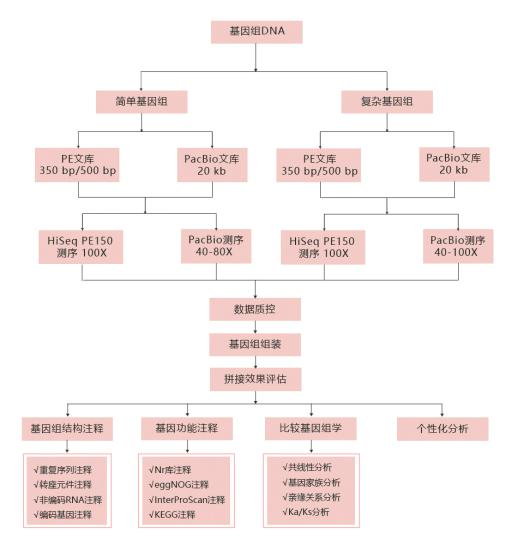
基因组从头测序(de novo sequencing),主要针对基因组序列未知或参考基因组组装不理想的物种,构建不同类型的基因组DNA文库,并进行序列测定。然后使用生物信息学方法对序列进行拼接、组装和注释,从而绘制该物种完整的基因组序列信息。简单基因组:重复序列低于50%,且二倍体杂合度低于0.5%的物种。复杂基因组:重复序列高于50%,或二倍体杂合度高于0.5%,或其他多倍体物种。
技术流程:
分析内容:
1.下机数据统计 | 11.杂合度分析 |
2.基因组组装效果评价 | 12.共有特有基因家族分析 |
3.蛋白编码的基因序列比对 | 13.Survey分析 |
4.全基因组复制事件分析 | 14.非编码RNA预测 |
5.数据质控 | 15.染色体共线性分析 |
6.基因组组装完整性与连续性评估 | 16.LTR插入时间 |
7.基因组注释 | 17.基因组组装 |
8.分歧时间估算 | 18.蛋白编码基因预测 |
9.高质量数据获取 | 19.基因家族扩张和收缩分析 |
10.重复序列分析 | 20.正选择分析 |
结果展示:




动植物全基因组重测序
动植物全基因组重测序是指在一个物种的基因组已知的情况,对该物种的大量样本进行测序。将得到的序列信息和参考基因组比对,得到该物种的全基因组遗传变异多态性,并进行群体水平的分析和统计,进行功能基因的定位或对该物种的遗传进化关系做出预测分析,估计生物种类的分化时间和速度等。
实验流程:
DNA提取→DNA 检测→PE150文库构建→文库质检→BGISeq或Illumina PE150测序→数据分析
分析内容:
测序数据及其质量控制 | 数据统计 |
二代测序数据及其质量控制 | |
测序数据产出统计 | |
比对 | 与参考基因组的比对统计 |
SNP calling | |
Indel检测,统计及注释 | |
SV/CNV统计,检测,注释 | |
群体分析(GWAS) | 群体进化树分析 |
群体结构分析 | |
群体主成分分析(PCA) | |
候选基因的筛选 | |
候选基因注释 | |
群体进化分析 | 群体进化树分析 |
群体结构分析 | |
群体主成分分析 | |
连锁不平衡分析(LD) | |
多态性分析、选择分析 | |
选择区域定位及基因注释 |
结果展示:


真菌基因组
真菌属于较低等的真核生物,种类繁多,在自然界中分布广泛。据估计,全世界约有150万种真菌,其中包含许多具有重要药用价值和食用价值的有益真菌,同时也存在着大量能引发动植物病害的致病菌。因此,合理利用有益真菌,控制和预防有害真菌对人类的生产和生活具有重要的意义。而真菌基因组的阐明,将为真菌特性的分析提供重要依据。随着高通量测序技术的成熟,基因组测序成本已大大降低,使得快速而经济的进行真菌基因组测序成为可能,目前真菌基因组学研究已经进入了一个快速增长时期。 目前通过Illumina和Pacbio/ONT平台测序并进行de novo基因组组装,即可获得获得真菌的基因组精细图并进行深度生物信息挖掘。
实验流程:
DNA提取→DNA 检测→Pacbio/ONT文库构建→文库质检→Pacbio/ONT测序
分析内容:
基因组组装 | 数据质控 |
基因组评估 | |
基因组组装 | |
基因分析 | 编码基因预测 |
重复序列注释 | |
非编码RNA预测 | |
假基因分析 | |
功能注释 | GO,KEGG,COG,Swiss-prot,Nr,pfam,CAZyme,PHI,信号肽预测,跨膜蛋白,转运蛋白,分泌蛋白分析, |
比较基因组 | 基因家族分析,特有共有基因检测,进化关系分析,共线性分析 |
结果展示:


细菌基因组
细菌全基因组从头测序(Bacterial Whole Genome de novo Sequencing ),是指对基因组序列未知或无近缘物种基因组信息的某个物种,构建不同插入片段的基因组DNA文库并对文库进行序列测定,然后利用生物信息学方法进行拼接、组装和注释,从而获得该细菌的基因组序列图谱。
分析内容:
基因组组装 | 基因组组装及结果评估 |
基因组结构研究 | 编码/非编码基因预测 |
重复序列分析 | |
基因岛预测 | |
前噬菌体预测 | |
CRISPR 预测 | |
基因基本功能注释 | 常见功能数据库注释 (NR、GO、COG、KEGG、SwissProt、Pfam、TCDB、CAZy) |
结果展示:

细菌完成图
细菌的染色体基因组通常仅由一条环状双链DNA分子组成细菌的染色体相对聚集在一起,形成一个较为致密的区域,称为类核(nucleoid)。细菌基因组学是研究细菌全基因组DNA 序列及其结构与功能的学科。1995 年, 科学家获得了流感嗜血杆菌(Haemophilus influenzae Rd)的全基因组序列, 这是第一个完整的基因组序列, 也是第一个完成的细菌基因组序列。随着高通量测序技术和生物信息学的快速发展,细菌全基因组de novo测序已然成为一种探究细菌生物学问题的高性价比方法。通过三代测序测序,可以获知待测菌株的基因及相关调控信息,为研究该菌株特有的生物学特征(致病机制,共生机制,独有的代谢机制)提供分子生物学基础;通过比较基因组分析,为研究菌株种内及种间的功能差异、进化关系提供理论指导。
实验流程:
DNA提取→DNA 样品检测→Pacbio/ONT文库构建→文库质检
分析内容:
1.数据污染评估 | 5.基因组结构注释 |
2.基因组组装 | 6.基因组功能注释 |
3.质粒分析 | 7.基因组圈图 |
4.基因组注释 |
结果展示:








叶绿体基因组
叶绿体基因组在很多方面与线粒体基因组的结构是相似的。每个叶绿体中cpDNA的拷贝数随着物种的不同而不同。但都是多拷贝的。这些拷贝位于类核区。叶绿体(cp)基因组测序是利用二代测序平台,对植物叶绿体基因组进行高通量测序,突破传统cpDNA分离提纯的实验壁垒,可直接利用组织总DNA获得叶绿体基因组,对序列信息进行深入解析,通过比较基因组分析获得物种分类、系统进化、地理谱系遗传等信息。
实验流程:
DNA提取→DNA 检测→Illumina 文库构建→文库质检→Illumina PE150测序
分析内容:
测序数据及其质量控制 | 测序碱基质量值 |
测序碱基含量分布 | |
测序质量控制 | |
测序数据产出统计 | |
基因组组装 | 拼接质量评估 |
基因预测结果统计 | |
基因组结构图 |
结果展示:




线粒体基因组
线粒体(mt)基因组测序是利用二代测序平台,对动植物线粒体基因组进行高通量测序,突破传统mtDNA分离提纯的实验壁垒,可直接利用组织总DNA获得线粒体基因组,对序列信息进行深入解析,通过比较基因组分析获得物种分类、系统进化、地理谱系遗传等信息。
实验流程:
DNA提取→DNA 检测→Illumina 文库构建→文库质检→Illumina PE150测序
分析内容:
测序数据及其质量控制 | 测序碱基质量值 |
测序碱基含量分布 | |
测序质量控制 | |
测序数据产出统计 | |
基因组组装 | 拼接质量评估 |
基因预测结果统计 | |
基因组结构图 |
结果展示:


病毒基因组
病毒基因组测序包括完成图测序、扫描图测序,通过二代/三代测序平台,获得病毒的基因组的序列信息,并在结构基因组学、比较基因组学层面通过差异分析、同源基因分析、共线性分析、物种进化分析等手段探究病毒的毒力系统、基因组的进化与演变历程等。对于纯化后的病毒,我们通过三代测序可以100%获得该病毒的完整基因组序列。
实验流程:
DNA提取→DNA 检测→PE文库构建→文库质检→ PE150测序
分析内容:
测序数据及其质量控制 | PacBio数据统计 |
二代测序数据及其质量控制 | |
测序质量控制 | |
测序数据产出统计 | |
序列组装 | 拼接质量评估 |
病毒基因组注释 | |
病毒基因组结构图 |
结果展示:




 技术咨询:
技术咨询: