整合单细胞与批量RNA测序:揭示卵巢癌中乳酸化调控代谢重编程与化疗耐药的关联
2025-12-02
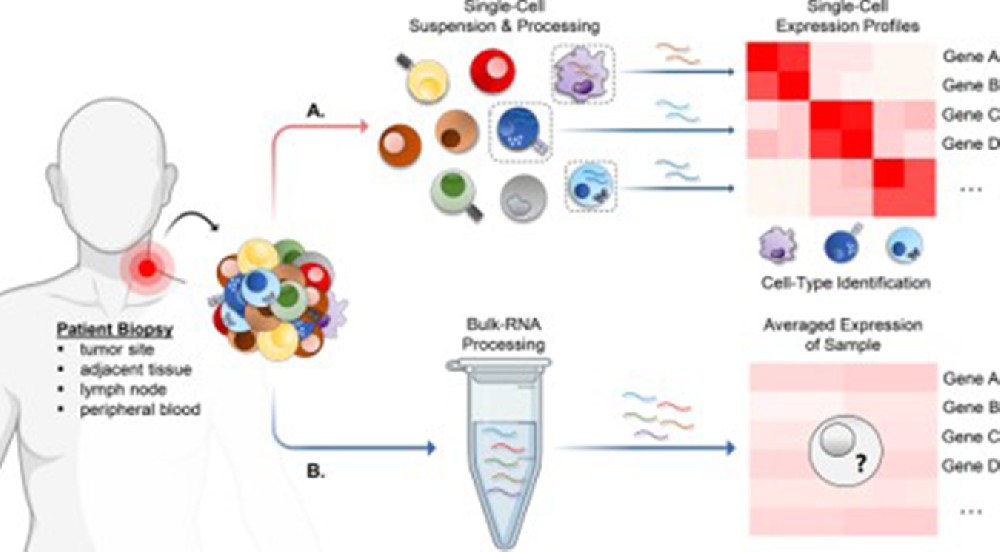
卵巢癌是女性生殖系统中最常见的恶性肿瘤之一,因其早期症状隐匿、易复发和化疗耐药性强,成为妇科肿瘤治疗的主要挑战。据统计,超过70%的晚期卵巢癌患者会对铂类化疗药物产生耐药,导致治疗失败和预后不良。近年来,代谢重编程被认定为癌症的核心特征之一,其中乳酸化作为一种新兴的代谢调控机制,逐渐引起研究者的关注。乳酸化是一种组蛋白修饰过程,通过乳酸代谢产物影响基因表达,进而调控细胞行为。在卵巢癌中,乳酸化可能与肿瘤微环境的改变和化疗耐药密切相关。
为了深入探索这一机制,研究人员结合了批量RNA测序(bulk RNA-seq)和单细胞RNA测序(scRNA-seq)技术,开展了一项创新性研究。这两种技术各有优势:bulk RNA-seq能够提供组织样本的整体基因表达谱,帮助识别在肿瘤与正常组织间的宏观差异;而scRNA-seq则能解析单个细胞的表达特征,揭示肿瘤异质性和细胞亚群中的细微变化。通过整合这两种方法,研究旨在精准定位与乳酸化相关的关键基因,并阐明它们在卵巢癌化疗耐药中的作用。本文将以一篇题为“Integration of scRNA-seq and bulk RNA-seq to reveal the association and potential molecular mechanisms of metabolic reprogramming regulated by lactylation and chemotherapy resistance in ovarian cancer”的学术论文为例,详细解析其研究思路、发现及临床意义,帮助临床医生和老师理解这一前沿领域。
背景:卵巢癌、代谢重编程与RNA测序技术
卵巢癌的发病机制复杂,涉及基因突变、表观遗传改变和代谢异常。代谢重编程是指癌细胞通过改变能量代谢途径(如增强糖酵解)来支持其快速增殖和生存,这一过程与化疗耐药密切相关。乳酸化是近年来发现的一种代谢相关修饰,它通过乳酸分子与组蛋白结合,调控基因转录,进而影响细胞分化、免疫应答和药物反应。在肿瘤微环境中,乳酸水平升高可能促进乳酸化,从而驱动癌症进展。
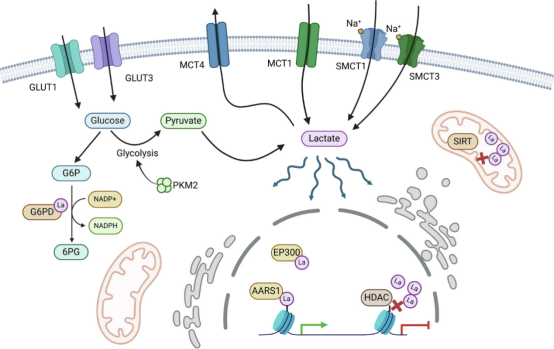
图1. 乳酸化修饰的机制
RNA测序技术是研究基因表达的重要工具。批量RNA-seq通过分析整个组织样本的RNA,能够快速识别在疾病状态下差异表达的基因,适用于大规模筛选。然而,它无法区分样本中不同细胞类型的贡献,这可能掩盖关键信息。例如,在卵巢癌中,肿瘤组织包含癌细胞、免疫细胞和基质细胞等多种成分,它们的基因表达模式各异。单细胞RNA-seq则弥补了这一不足,通过对单个细胞进行测序,可以揭示细胞亚群的特异性表达,帮助识别驱动耐药的稀有细胞类型。
整合这两种技术,能够从宏观和微观层面全面解析基因表达,为识别可靠的生物标志物和治疗靶点提供坚实基础。本研究正是基于这一思路,聚焦于乳酸化相关基因,探索它们在卵巢癌耐药中的作用。
研究思路解析:从大数据筛选到精准定位
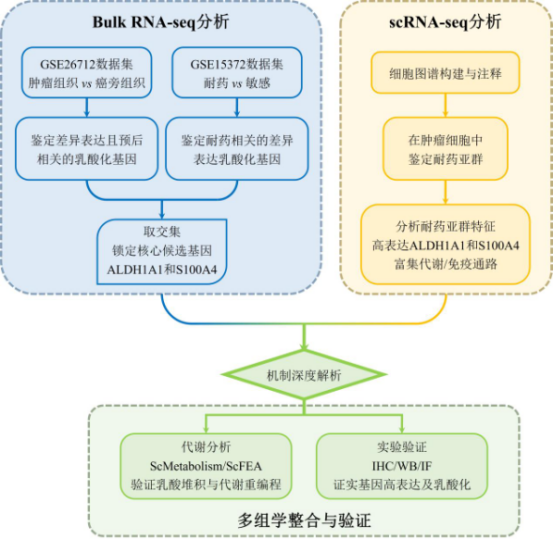
图2. 研究思路解析
该研究采用三步法,逐步缩小范围,最终锁定与乳酸化调控和化疗耐药相关的关键基因。整个过程类似于“侦探破案”,通过多重证据链确保结果的可靠性。
第一步:初步筛选——识别在肿瘤中异常表达且与预后相关的乳酸化基因
研究首先利用卵巢癌的批量RNA-seq数据,比较了肿瘤组织与正常组织之间的基因表达差异。重点关注乳酸化相关基因,这些基因通常参与代谢途径或组蛋白修饰。通过统计方法,如差异表达分析,计算基因的表达变化倍数和显著性p值,筛选出在肿瘤中显著上调或下调的乳酸化基因。同时,结合患者生存数据,分析这些基因与预后的关联,例如,高表达某基因是否与患者总生存期缩短相关。
这一步的目的是从大量基因中初步筛选出可能在卵巢癌发展中起作用的“嫌疑基因”。例如,某些乳酸化基因可能在肿瘤组织中过度活跃,驱动细胞增殖或抑制凋亡,从而导致预后不良。通过这种差异表达和预后关联的双重筛选,研究确保了所选基因不仅与癌症相关,还具有临床意义。
第二步:聚焦耐药——在化疗耐药样本中进一步筛选基因
接下来,研究转向另一套批量RNA-seq数据集,该数据集来源于对化疗耐药与敏感卵巢癌样本的比较。化疗耐药是卵巢治疗中的核心问题,通常与基因表达改变有关。在这一步中,研究人员分析了耐药组与敏感组之间差异表达的乳酸化基因,筛选出在耐药样本中显著变化的基因。
这一步骤直接关联临床实践,因为识别耐药相关基因有助于预测患者对治疗的反应。例如,某些基因可能在耐药样本中高表达,可能通过促进药物外排或增强DNA修复机制,导致化疗失效。通过这一步,研究进一步缩小了范围,聚焦于那些既在肿瘤中异常表达,又在耐药中发挥作用的基因。
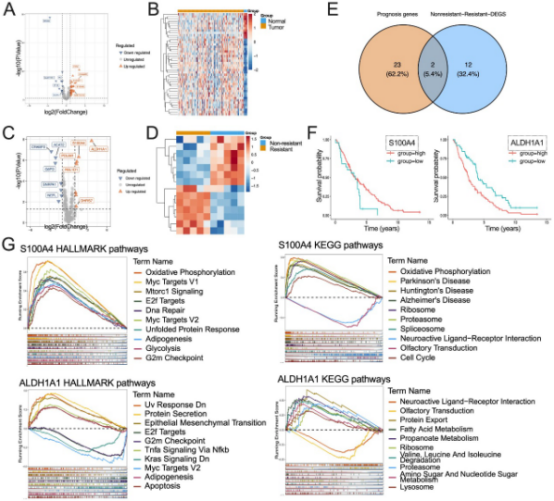
图3. 第一步与第二步的筛选结果
第三步:锁定关键——取交集鉴定潜在耐药基因
最后,研究将前两步得到的基因集取交集,即筛选出同时满足三个条件的基因:在肿瘤中差异表达、与患者预后相关,且在耐药组中差异表达。这种严格的筛选方法大大降低了假阳性风险,确保了结果的精准性。通过这一过程,研究成功鉴定出两个关键基因:ALDH1A1和S100A4。
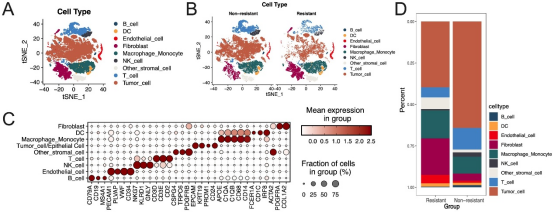
图4. 第三步的鉴定结果
ALDH1A1是醛脱氢酶家族成员,参与细胞解毒和干细胞特性维持,在多种癌症中与化疗耐药和复发相关。S100A4是一种钙结合蛋白,涉及细胞运动、侵袭和转移,其过表达常预示不良预后。这两个基因均与乳酸化调控的代谢重编程有关,可能通过影响能量代谢和表观遗传修饰,促进卵巢癌的耐药性。
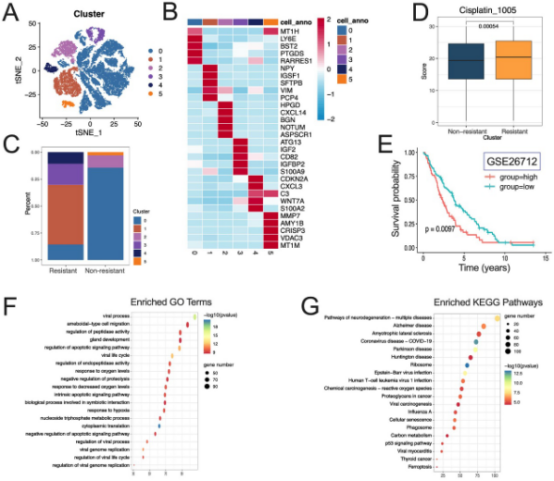
图5. 单细胞RNA测序与批量RNA测序联合分析
实验验证:从生物信息学到功能证实
生物信息学分析为研究提供了重要线索,最终结论需要通过实验验证。本研究后续采用多种实验方法,证实了ALDH1A1和S100A4在卵巢癌耐药中的功能。
1. 免疫组化(IHC):通过在卵巢癌组织切片中检测ALDH1A1和S100A4的蛋白表达,验证了它们在肿瘤样本中的高表达,且与患者临床特征(如肿瘤分级和耐药性)相关。这为生物信息学预测提供了形态学证据。
2. 细胞培养实验:利用卵巢癌细胞系,研究人员通过基因敲低或过表达技术,改变ALDH1A1和S100A4的表达水平。结果发现,敲低这些基因后,癌细胞对化疗药物的敏感性增强,而过表达则导致耐药性上升。这表明这两个基因直接参与调控化疗反应。
3. 免疫荧光:通过可视化技术,显示ALDH1A1和S100A4在细胞中的定位,进一步阐明了它们可能的作用机制。
这些实验不仅验证了生物信息学预测的准确性,还深入揭示了基因的功能机制。
临床意义与未来展望
这项研究的发现对卵巢癌的临床诊疗具有重要启示。首先,ALDH1A1和S100A4可作为潜在的生物标志物,用于预测患者对化疗的反应和预后。通过检测这些基因在肿瘤组织中的表达水平,医生可能提前识别耐药高风险患者,从而调整治疗方案。其次,这些基因及其相关通路可能成为新的治疗靶点。此外,乳酸化调控机制的揭示,开辟了代谢靶向治疗的新领域。通过干预乳酸代谢或乳酸化过程,可能逆转耐药性,提高化疗效果。
对于临床医生和老师而言,理解这类多组学整合研究的意义在于,它强调了基础研究与临床实践的紧密结合。生物信息学方法能够快速筛选大量数据,提供候选靶点,而实验验证则确保这些靶点的可靠性。未来,随着技术的进步,类似研究有望应用于更多癌症类型,推动精准医学的发展。
结论
研究的主要筛选步骤依赖于批量RNA-seq,但单细胞RNA-seq在后续分析中发挥了关键作用。通过scRNA-seq,研究人员能够解析卵巢癌肿瘤微环境中的细胞异质性。这种单细胞水平的分析,帮助揭示了耐药性的细胞起源,为开发靶向疗法提供了新方向。整合scRNA-seq和bulk RNA-seq的优势在于,它既提供了整体视角,又揭示了细节差异。在临床应用中,这意味着可以更精准地识别高危患者亚群,并设计个性化治疗策略。
参考文献:Ren F, Pang X, Jin F, Luan N, Guo H, Zhu L. Integration of scRNA-seq and bulk RNA-seq to reveal the association and potential molecular mechanisms of metabolic reprogramming regulated by lactylation and chemotherapy resistance in ovarian cancer. Front Immunol. 2025;16:1513806. DOI: 10.3389/fimmu.2025.1513806
【本研究通过对公共数据库(包括205例Bulk RNA-seq与13例scRNA-seq数据)的深入挖掘,揭示了卵巢癌代谢与耐药机制方面的重要线索。若从头开展实验获取同等规模原始数据,仅双组学检测与联合分析环节的成本约需24万元(不含其他实验开支)。
在实际科研中,我们理解样本量并非越大越好,关键在于能否充分支撑科学结论。针对不同课题的具体目标与研究设计,所需Bulk RNA-seq与scRNA-seq的样本数量往往可显著优化—无需盲目追求大样本量,而应围绕核心科学问题,合理确定能够严谨验证假说的最小有效样本规模。
我们可为老师提供专业的实验设计支持,结合课题关注的关键靶点与通路,评估并推荐更具成本效益的Bulk与单细胞RNA测序样本数量,协助制定样本更精简、预算更集中的靶向验证方案,从而以最优投入获取充分、可靠的科研结论,最大化科研资源的使用效率。】
 技术咨询:
技术咨询: