
原文链接:中药网络药理学完全攻略V4.0|分子对接(下):PyMOL可视化与结果解读!
导入:
网络药理学研究做到这里,大家是不是也有同样的疑问:
成分和靶点确定了,能不能再“看一眼”,它们到底是怎么结合的?
答案就是——分子对接!
网络药理学完全攻略V4.0来了,这次我们带你走进分子对接的世界,从零开始,连更三期,手把手搞定整个流程!
对接前的准备是重中之重,本篇我们就从最基础的软件配置开始,带你一步步走过:
软件准备与下载路径
小分子配体、蛋白受体的获取
数据预处理与格式转换
别担心,看似繁琐,其实只要理清逻辑,操作起来顺滑得像滑冰!

一、软件准备和下载路径
1.分子对接所需要软件
Autodock/Autodock Vnia--------分子对接的核心组件
Chem3D-------分子结构准备与构建的处理平台(在小分子结构无3D结构时可转化)
Open Bable GUI------多种格式转化的必备工具
PyMOL-----分子/蛋白处理及可视化的得力助手
概而言之:AutoDock/Vina 是负责核心计算的“引擎”, Chem3D 是重要的“准备车间”和“辅助观察窗”,Open Bable GUI是“万能适配器”和“高效搬运工”,而 PyMOL 则是最终的“精密检测仪”、“高清显示屏”。
小提示:在安装软件时,务必根据操作系统选择对应版本,以上软件可去官网下载适合自己电脑配置的版本,也可进行网上购买安装教程。
2.软件下载
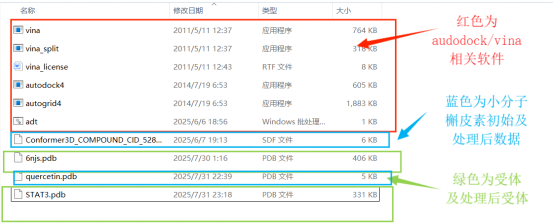
新建文件夹,将下载的Autodock和后期所用的小分子和受体统一到一个文件夹。
注意事项:
使用Autodock的组件必须将其与小分子和蛋白受体放置到同一文件夹中
文件夹及文件夹内内容命名均不可出现中文和空格,不然会导致报错!!
二、小分子配体和蛋白受体的下载
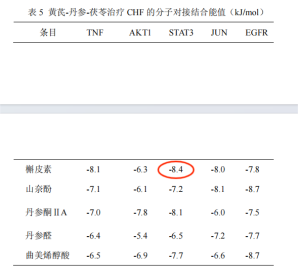
如文章中表5所示槲皮素-STAT3的结合能最高,为-8.4 kJ/mol
因此我们选择使用Autodock Vina复现槲皮素(Quercetin)与STAT3间的分子对接过程。其中槲皮素为小分子化合物配体,而TNF为蛋白受体。
1. 小分子配体的准备
①打开Pubchem网站:https://pubchem.ncbi.nlm.nih.gov/
输入槲皮素的英文quercetin,进行查询
Tips:通常BEST MATCT就是我们要寻找的最佳目标,不过也要注意甄别筛选。
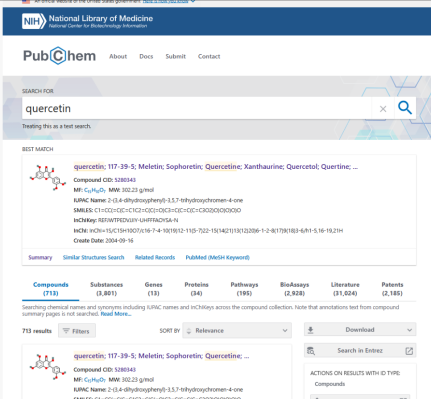
②选择进入后 我们能够看到槲皮素(quercetin)的一些基本信息及2D/3D结构图,选择右上角的Download。
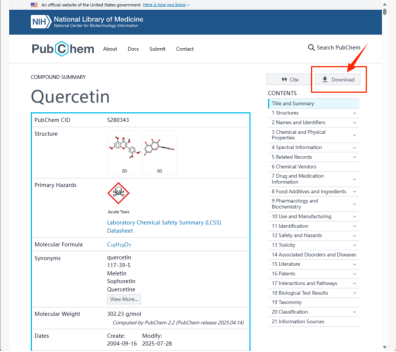
③在目标分子配体有3D结构的情况下,我们优先选择3D结构的SDF格式进行下载
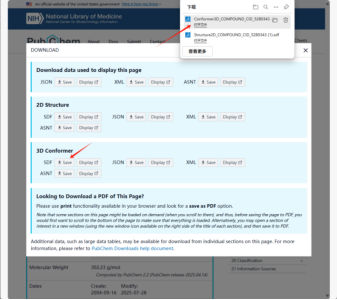
2. 蛋白受体准备
①选择UNIPROT数据库https://www.uniprot.org进行查询
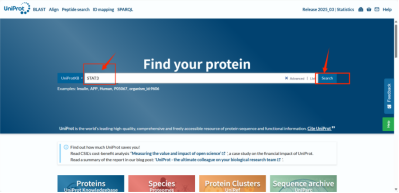
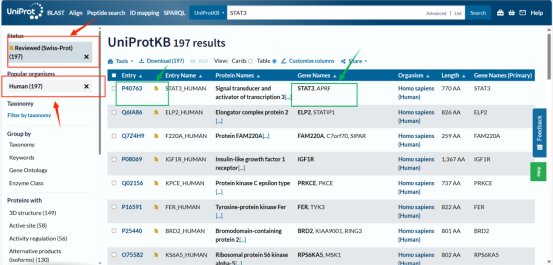
Tips:在输入STAT3后我们会查询到诸多结果。首先我们要在目录列表左侧进行初筛(下图红色箭头标注)选择Reviewed(已审核)和Human(人类)两个选项,使结果更具有真实性和可靠性。然后我们检查Gene Names选项,确认其为STAT3后,记住其Entry编号(p40763)一般来说初筛后第一个就是我们的目标受体(下图绿色箭头标注)。
②然后我们选择PDB数据库http//:www2.rcsb.org,输入STAT3的Entyr编号
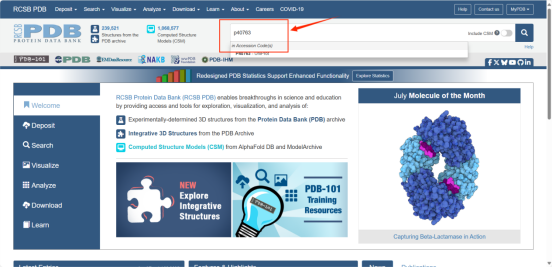
Tips:因为蛋白受体结构的提取和检测的方法不一,所以PDB数据库一般会提供多个受体选择,我们可根据自己情况适当选择
选择优先级原则:
1.(分辨率(Resolution)
优先选择分辨率最高的结构(数值越小越好)。
≤ 2.0 A:理想选择,原子位置精确度高。
2.0 - 2.5 A:可接受,需检查电子密度图(若可用)。
> 2.5 A:谨慎使用,可能影响对接准确性。
2.与目标研究区域的匹配度
STAT3的关键功能域是 SH2结构域(磷酸化酪氨酸 pTyr705 结合位点)。
确认结构包含完整的SH2结构域(残基约 580-670)。
若研究蛋白-蛋白相互作用(如二聚化),选择包含 DNA结合域(DBD) 和 连接域 的全长/近全长结构。
3.配体结合状态(Ligand Presence)
有抑制剂/配体共结晶的结构:优先选择(尤其当配体与你的化合物类似时)。
*例如:STAT3抑制剂(如 STX-0119, Stattic, S3I-201)复合物结构。*
无配体(Apo)结构:适合全新位点筛选,但需注意构象变化。
4.生物组装状态(Biological Assembly)
STAT3的活性形式是 磷酸化二聚体(pTyr705)。
选择 磷酸化(pTyr705)状态的结构(检查PDB注释中的"Modified Residue")。
确认结构是否为 生理二聚体(查看PDB中的"Biological Assembly")。
5.人源(Homo sapiens):首选(UniProt编号:P40763)。其他物种(如小鼠):仅在保守性高时备用(需序列比对验证)。
因此我们选择第一个结构,因为他的分辨率最高,为2.7A
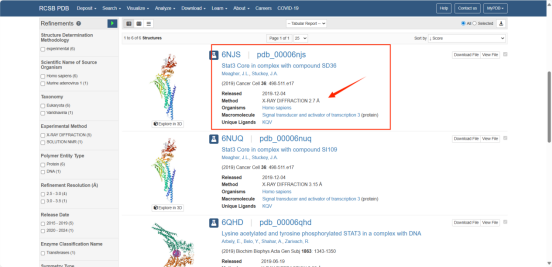
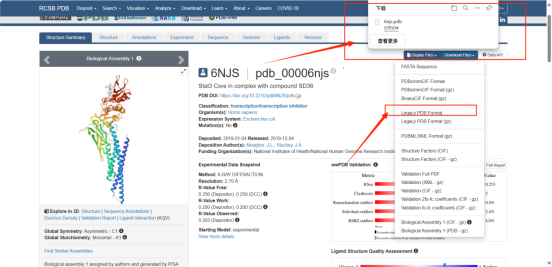
③选择第一个蛋白结构后,我们点击右上角的Download Files,选择Legacy PDB Format格式进行下载。
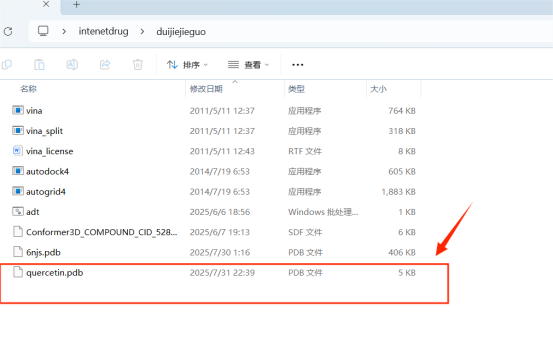
④至此我们已经拥有了小分子配合和蛋白受体的基本结构文件(如下图所示)
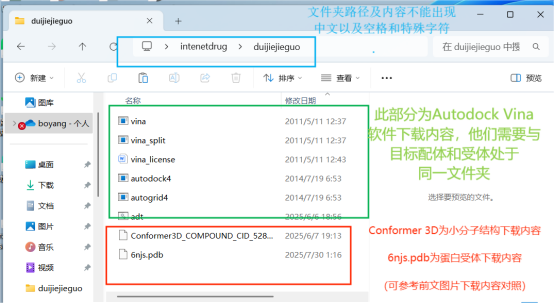
三、小分子配体和蛋白受体加工处理
1.小分子格式转换
首先使用Open Bubal GUI软件将SDF格式小分子配体转换为PDB格式;
导入和导出文件格式分别选择SDF和PDB格式;
设置导入文件夹和到处文件夹路径(应和前文文件夹一致);
点击CONVERT,等待结果导出。
具体操作步骤见下图:
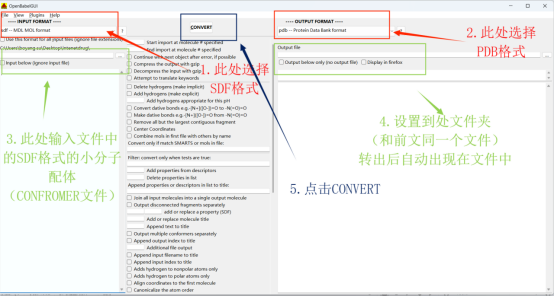
转换完成后,我们发现原本的SDF文件(Conformer3D文件)已经转换为PDB格式的文件了(重命名为quercetin,槲皮素),至此我们已经拥有了STAT3(6njs)和槲皮素(quercetin)的PDB格式的文件
2. PyMOL对受体进行处理
①打开PyMOL将STAT3受体文件拖入PyMOL
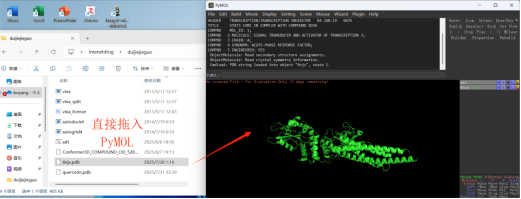
此时我们将STAT3已经能够看到STAT3受体的结构了,但是能够观察到受体周围有很多水分子(白色箭头处的红色点点)和多余的小分子结构(黄色箭头区域)
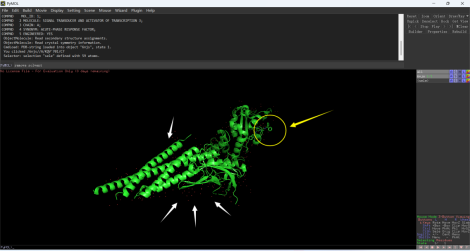
②在下图黄色区域内分别输入指令remove solvent 和remove organic去除多以水分子和小分子(分别输入后,分别按ENTER键确认)
Tips:处理后的蛋白结构更干净,对接结果更准确
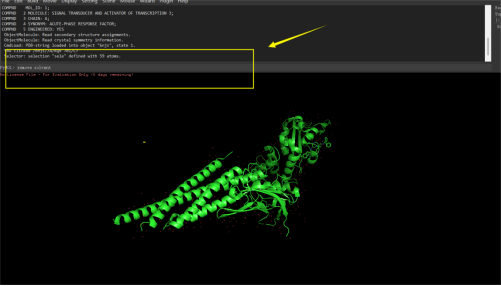
③将处理好的受体文件进行保存:点击左上角File→Export Molecule→save→选择PDB格式保存(此处我命名为stat3,同样保存到同一文件夹中)
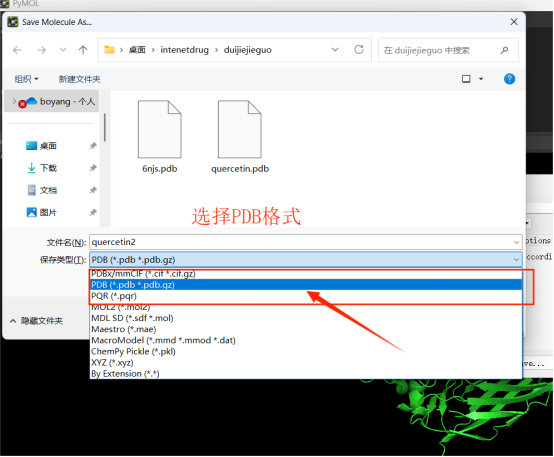
四、小分子配体和受体加氢处理及PDBQT格式转化
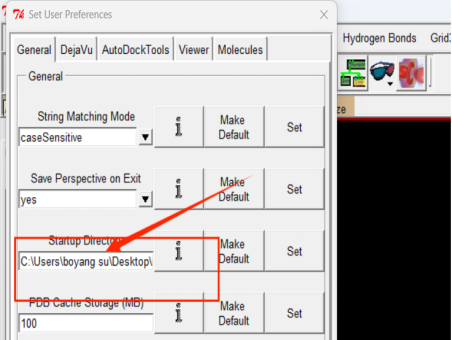
1.Autodock设置默认文件路径
左上角File→preference→set→将路径设置为之前的文件夹(文件路径不能有中文和空格)
目前文件夹内内容展示
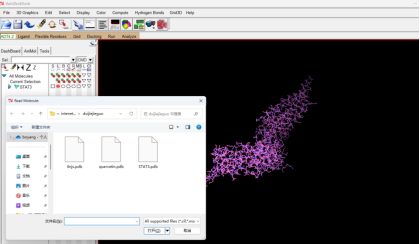
2. 受体pdbqt格式转化
①受体选择
File→read molecule→选择STAT3(如下图)
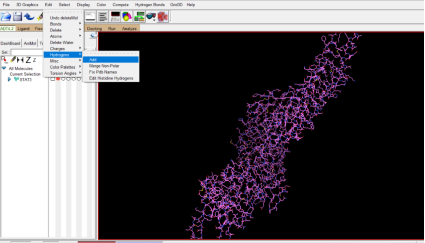
②受体加氢
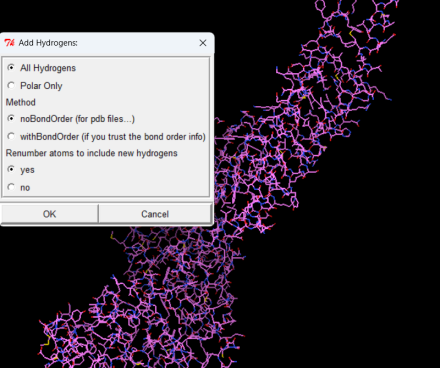
Edit→Hydrogens→add→ok
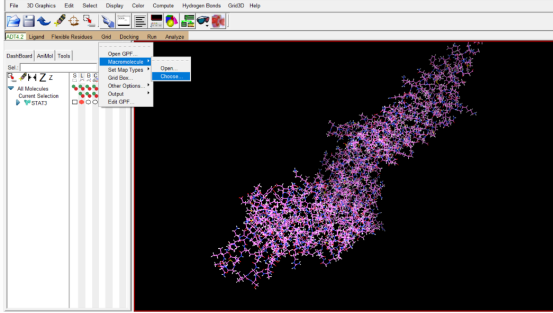
③将STAT3选为受体及PDBQT格式转化
Grid→Macromolecule→choose→STAT3→Select Molecule→命名为STAT3-pdbqt(一定要以pdbqt形式保存,如果未成功保存可自行更改文件名后缀,依然保存到上述文件夹中
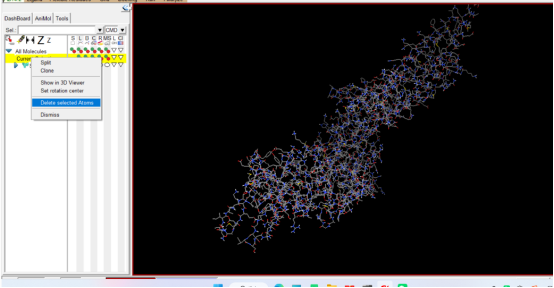
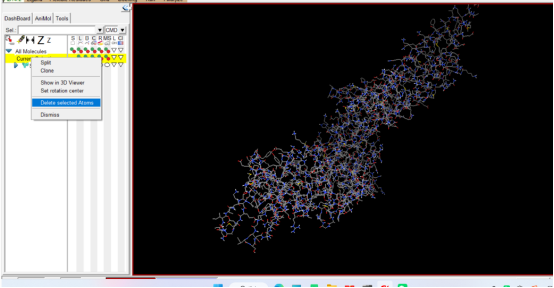
④删除受体文件文件→下一步进行小分子处理(鼠标左键选择current...鼠标右键→Delete selected Atoms)
3.小分子pdbqt格式转化
①打开小分子
Ligan→input→open→选择槲皮素(querceine,pdb格式)
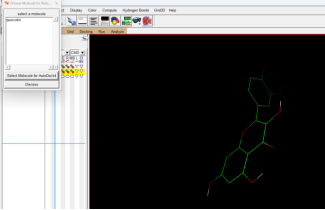
② 将槲皮素作为配体并检测扭转
按照如下步骤操作,将槲皮素作为配体,并初步观察小分子扭转和结合点
Ligant→In put→choose→选择槲皮素→Select Molecule for Autodock
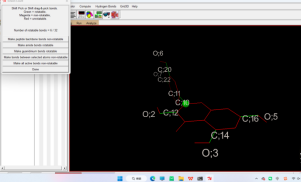
Ligant→Torsion tree→Detect Root
Ligant→Torsion tree→Choose Torsion...
(如下图,绿色代表可自由旋转,红色不可以自由旋转,粉红色则代表两者之间按住shift点击粉红部分则可将其变为可扭转部分,若不处理则默认为不可扭转部分)
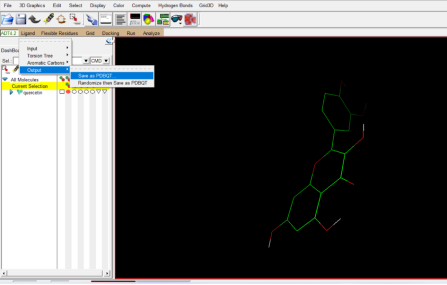
③小分子pdbqt格式转化
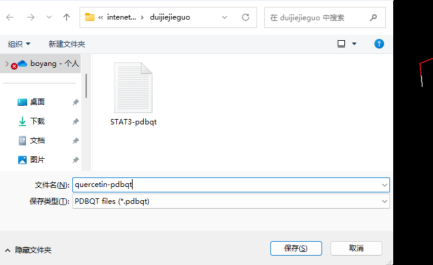
Ligant→Out put→Save as PDBQT重命名为quercetin-pdbqt→删除配体
五、小结
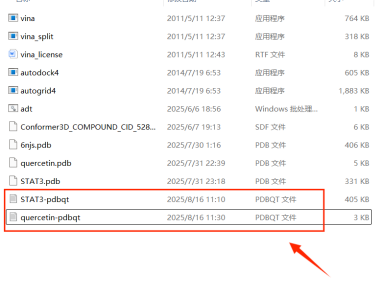
至此,我们拥有了受体(STAT3-pdbqt)和小分子配体(quercetin-pdbqt)的PDBQT格式文件,(下图红色箭头标记部分)已完成全部分子对接准备步骤,下一步,就可以用 AutoDock Vina 开始分子对接实战啦!
今天我们完成了分子对接的第一步:软件准备 + 数据下载与预处理。
是不是觉得步骤不少,但其实流程很清晰?
下篇预告:
我们就要正式进入 AutoDock Vina 分子对接实操,带你跑通一个完整对接流程!
 技术咨询:
技术咨询: