益生菌卷到肝了!植物乳杆菌靠一条lncRNA,让肝脏自噬"卷"起来抵御药物性肝损伤
2026-07-09

益生菌卷到肝了!植物乳杆菌靠一条lncRNA,让肝脏自噬"卷"起来抵御药物性肝损伤
肠菌代谢物HMB遥控肝脏lncRNA,抵御药物性肝损伤
浙江大学转化医学研究院/浙江大学医学院附属第一医院王宇浩研究员团队通过RNA-pull down、RNA-seq、双荧光素酶报告基因实验、16S rRNA测序、粪菌移植、代谢组学等技术,揭示了肠道有益菌通过代谢产物2-羟基-2-甲基丁酸(HMB)上调肝脏lncRNA SNHG9表达,进而激活“IMP2-MYC-MAS”信号轴并引发肝细胞保护性自噬,从而缓解对乙酰氨基酚(APAP)诱导的药物性肝损伤,为药物性肝损伤的预防和治疗提供了新的干预策略。
文献标题:Hepatic SNHG9 links gut microbiota to liver protection in drug-induced liver injury
文献来源:王宇浩/卢言慧/潘金水/陈佳欣共同通讯
发表期刊:Nature Communications(IF=15.7,JCR Q1区)
发表时间:2026年5月
文章链接:https://doi.org/10.1038/s41467-026-73309-4
研究背景:
药物性肝损伤(Drug-induced liver injury, DILI)是急性肝衰竭的重要诱因之一,对公共健康构成显著威胁。对乙酰氨基酚(Acetaminophen, APAP)过量服用是全球DILI的常见诱因,而目前DILI的治疗选择有限,亟需探索新的治疗策略。近几年已有报道揭示DILI受到肠道菌群及其活性物质的调节,一些菌群代谢物在DILI中起到保护作用。前期研究发现,长链非编码RNA(lncRNA)SNHG9是肠道中关键宿主分子,也在肝脏中表达,但其在肝-肠轴通信及DILI中的作用尚不明确。
关键研究发现
1、肝脏SNHG9可缓解APAP诱导的肝损伤
肠道菌群调控SNHG9表达:不同饲养环境的两组小鼠肝脏SNHG9表达差异显著(图1A-C);粪菌移植(FMT)证实菌群直接调控SNHG9。
APAP诱导SNHG9动态变化:APAP处理后,小鼠肝脏SNHG9表达量早期下降,24h显著升高,提示SNHG9在肝毒性应激下受到动态调控(图1D)。
SNHG9过表达保护作用:与对照组相比,SNHG9过表达小鼠的APAP损伤减轻,表现为丙氨酸氨基转移酶(ALT)和天冬氨酸氨基转移酶(AST)水平均低于对照组(图1E-F),肝脏细胞凋亡减少(图1G),炎性因子(TNF-α、IL-6、IL-1β)表达下降(图1H-J)。荧光原位杂交(FISH)染色显示,DILI患者肝脏中SNHG9表达高于健康对照(图1K)。
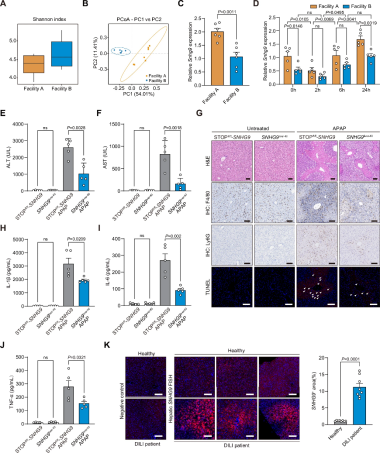
图1 肝脏SNHG9减轻对乙酰氨基酚所致肝损伤
2、SNHG9通过增强MAS介导的自噬缓解肝损伤
SNHG9调控MAS介导的自噬:RNA-seq显示Mas1(编码MAS)在SNHG9过表达小鼠中显著上调(图2A),qPCR、WB和免疫组化进一步印证了这一结果(图2B-D)。与转染非靶向对照sgRNA的编辑细胞相比,敲除SNHG9可使MAS表达水平降低,进而导致AKT磷酸化水平升高及FOXO1乙酰化水平降低,基础LC3-II水平降低并促进p62积累,但在敲除细胞中重新表达SNHG9可消除上述效应(图2E-F)。
SNHG9依赖MAS促进自噬:与敲除SNHG9的细胞相比,SNHG9过表达可上调MAS蛋白水平,AKT磷酸化水平降低、FOXO1乙酰化水平升高及自噬流改善,但加入MAS受体拮抗剂A779可消除上述效应(图2G-H),证实MAS是关键下游靶点。
自噬激活:与对照组相比,SNHG9过表达小鼠肝脏Mas1表达升高,MAS蛋白水平上调,FOXO1乙酰化增强,AKT磷酸化水平降低(图2I-J),且自噬相关指标(LC3-II、Ulk1、Atg7、Lamp1)表达均上调(图2K-L),自噬空泡数量显著增加(图2M),免疫荧光(IF)显示DILI患者肝脏MAS水平亦升高(图2N),提示SNHG9通过激活MAS依赖的自噬缓解APAP诱导的肝损伤。
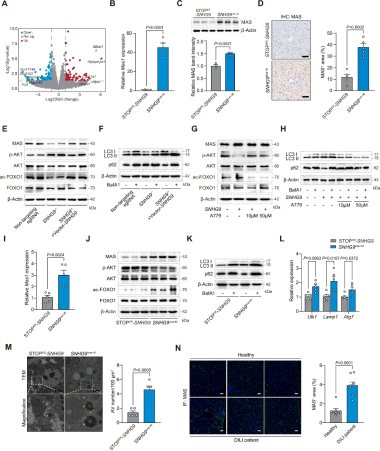
图2 SNHG9激活MAS介导的自噬,从而发挥肝保护作用
3、SNHG9可通过IMP2–MYC轴上调/下调MAS表达
SNHG9与IMP2结合:RNA-pull down和RIP证实SNHG9与IMP2存在直接互作(图3A-C)。为鉴定IMP2在SNHG9上的具体结合位点,构建了在不同环区缺失的SNHG9突变体并进行RNA-pull down,发现IMP2特异性结合SNHG9的环3区(图3D-E)。
IMP2抑制MYC翻译:RNA-seq显示,SNHG9过表达小鼠中MYC mRNA水平降低(图3F)。IMP2敲低可使MYC mRNA稳定表达并促进其翻译(图3G-H),而IMP2过表达的结果相反(图3K)。过表达SNHG9可增加IMP2与MYC mRNA的结合,而过表达SNHG9-D3则未显示类似效应(图3I),经APAP处理的SNHG9过表达小鼠肝脏MYC mRNA和蛋白水平均显著低于对照组(图3J-K)。这表明SNHG9与IMP2的结合可促进IMP2与MYC mRNA之间的互作。
MYC抑制MAS:Huh7细胞中MYC过表达可降低MAS的mRNA和蛋白水平(图3L-M),而MYC敲低则可升高其水平(图3N-O),表明MYC抑制MAS表达。HEK-293T细胞的双荧光素酶报告基因实验显示MYC可直接抑制MAS1启动子活性(图3P)。Huh7细胞的染色质免疫共沉淀-定量PCR(ChIP-qPCR)证实了MYC与MAS1启动子的直接结合(图3Q)。在稳定表达SNHG9的Huh7细胞中过表达MYC可显著逆转MAS的mRNA和蛋白水平(图3R、S)。上述发现表明MYC直接结合MAS1启动子抑制其转录,SNHG9通过抑制MYC解除转录抑制,上调MAS。
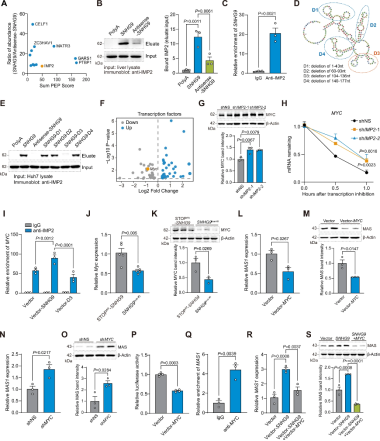
图3 SNHG9经IMP2–MYC轴调控MAS表达
4、乳杆菌富集激活SNHG9–MAS信号通路并对抗APAP诱导的肝损伤起保护作用
菌群筛选:结合图1结果,选取3种菌株对小鼠灌胃,发现只有植物乳杆菌(L. plantarum)显著上调肝脏SNHG9表达(图4A)。
L. plantarum可激活SNHG9–MAS信号通路:APAP诱导肝损伤小鼠灌胃L. plantarum可上调肝脏SNHG9表达(图4B),MYC的mRNA和蛋白水平均降低,MAS表达升高(图4C-D),MAS相关自噬信号增强,表现为AKT磷酸化降低、FOXO1乙酰化升高,以及Ulk1、Atg7和Lamp1表达上调(图4D-E),从而改善自噬流(图4F)。TEM进一步显示L. plantarum处理小鼠肝脏中自噬空泡增多(图4G),证实自噬程序被强烈激活。
L. plantarum可改善肝损伤:APAP诱导肝损伤小鼠灌胃L. plantarum 使24h血清ALT和AST水平降低(图4H),肝脏坏死减轻、Ly6G⁺中性粒细胞和F4/80⁺巨噬细胞浸润减少以及肝脏凋亡减少(图4I),血清促炎细胞因子水平降低(图4J)。
综上,L. plantarum可激活SNHG9–MAS信号通路,增强自噬,并减轻APAP所致肝损伤。
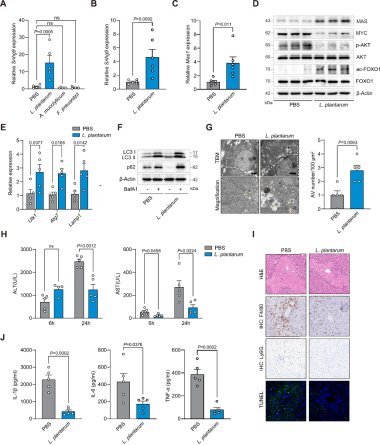
图4 乳杆菌富集激活SNHG9–MAS通路并减轻APAP所致肝损伤
HMB是关键代谢物:与灌胃新鲜培养基相比,小鼠灌胃灭活L. plantarum培养上清可有效诱导肝脏Snhg9表达(图5A),显著降低APAP所致肝毒性小鼠的血清ALT和AST水平(图5B),提示L. plantarum分泌的特定代谢物驱动SNHG9诱导表达及肝保护。L. plantarum培养上清及L. plantarum处理小鼠血清的准靶向代谢组学分析显示HMB在两种样本中均一致富集(图5C)。L. plantarum给药可升高小鼠循环HMB水平(图5D),人类血清样本中HMB浓度与粪便乳杆菌丰度呈正相关(图5E),与上述结果一致。同时,HMB给药可增加自噬空泡数量(图5F)。
HMB依赖SNHG9:为确定HMB介导的肝保护是否依赖SNHG9,构造肝脏Snhg9特异敲除小鼠模型,发现了Sngh9确实显著削弱了HMB的保护作用:与HMB处理的对照组小鼠相比,Sngh9敲除小鼠24h血清ALT和AST水平恢复正常(图5G),肝脏坏死加重、炎症细胞浸润增加、肝细胞凋亡增加(图5H),循环促炎细胞因子升高(图5I)。在DILI患者中观察到血清HMB水平与ALT和AST浓度均呈负相关(图5J)。综上,HMB可促进肝脏SNHG9表达并激活SNHG9–MAS信号通路,从而减轻APAP所致急性肝损伤。
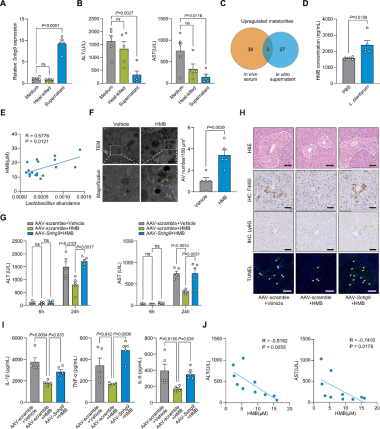
图5 乳杆菌经代谢物HMB激活SNHG9–MAS信号通路
6、HMB通过激活HCAR2诱导SNHG9表达
HCAR2是HMB受体:在Huh7细胞中沉默HCAR2可显著抑制SNHG9表达(图6A-B),表明HCAR2是HMB介导SNHG9上调所必需的。CRE-荧光素酶报告基因实验显示,HMB处理可显著抑制HCAR2调控的cAMP信号(图6C),微量热泳动(MST)分析证实了HMB与HCAR2的直接结合(KD≈66.7μM)(图6D)。表明HMB为HCAR2的功能性配体,并支持其在介导SNHG9诱导中的作用。
HCAR2为传递HMB信号的中间环节:HCAR2抑制剂MPN阻断HMB的保护作用,表现为MPN处理逆转了HMB诱导的肝脏Snhg9、Myc和Mas1表达变化(图6E–G),以及下游自噬信号标志物的变化(图6H),血清ALT和AST水平恢复至未接受HMB处理小鼠的水平(图6I、J),肝坏死、Ly6G⁺中性粒细胞和F4/80⁺巨噬细胞浸润以及肝细胞凋亡均与未接受HMB处理组相当(图6K、L)。综上,HMB通过HCAR2激活SNHG9–MAS信号通路。
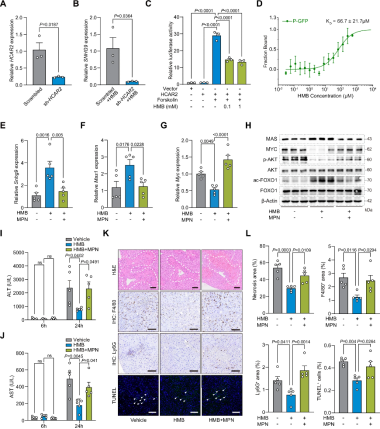
图6 HMB诱导的SNHG9–MAS信号激活依赖HCAR2
结论
综上所述,为阐明肠道菌群调控药物性肝损伤的肠-肝轴机制,该研究利用肝特异性基因编辑小鼠、细胞模型及人肝活检,聚焦长链非编码RNA SNHG9,首次发现并阐明了“HMB-SNHG9-MYC-MAS”这一保护性信号通路,揭示了肝脏SNHG9响应菌源性信号、启动细胞自噬以缓解DILI的新机制。同时也为临床通过补充植物乳杆菌等高表达HMB的益生菌进行DILI的早期干预与主动预防,提供了具有转化前景的新思路。
 技术咨询:
技术咨询: