从“一堆微生物”到“单个微生物”:单细胞转录组如何带来新发现?
2025-03-14
在我们眼中,微生物往往是“又多又杂”的存在:无论是肠道菌群、病原微生物还是环境中的菌群,都由无数不同种类和数量级的微生物构成。
然而,从这些“庞大群体”的表面现象去推测每个微生物的具体作用,难免失之粗糙。就好比站在远处看一场交响乐表演,只能听到整体的旋律,却无法分辨每个乐手的独特演奏技巧与音色。
微生物单细胞转录组测序(Microbial single-cell RNA sequencing, mscRNA-seq)的兴起为我们打开了新的研究视角。在该技术加持下,研究者可以“拆解”微生物群体至单个细胞,查看它们在转录水平上都做了些什么,从而深入解析那些容易被群体平均值掩盖的细节。尤其是在生物医学领域:从探究致病菌与宿主免疫反应的交互,到追踪细菌耐药性突变的产生,再到利用工业微生物生产药物和疫苗的优化过程,单细胞层面的“放大镜”正让我们更精准地理解微生物的功能与行为模式。
本篇文章将带你回顾mscRNA-seq在生物医学方向的最新进展。我们会聚焦几个具有代表性的研究领域,为你展现这项技术如何在医疗与健康领域不断拓宽边界,并感受到微生物世界的神奇和生命科学技术的无限可能。
1、高致病菌对宿主细胞的影响
Meng等人使用了一种高通量的跨物种双scRNA-seq技术,通过使用随机引物同时捕获真核生物和细菌的RNA(scRandom-seq)[1]。scRandom-seq可以高通量、高特异性地检测单个真核细胞和细菌细胞RNA表达谱。鲍曼不动杆菌(A .b)是一种高度致病菌和院内致病菌,对抗生素高度耐药,对人类健康造成重大威胁,亟待发现和治疗。
在A .b感染模型中,scRandom-seq发现了THP-1来源的巨噬细胞极化和细胞内A .b诱导宿主细胞铁死亡应激。Ferrostatin-1 (Fer-1)抑制铁死亡可提高细胞活力和抗A .b感染能力,表明其具有抗相关感染的潜在活性(图1)。scRandom-seq提供了一种高通量的跨物种双单细胞RNA谱分析工具,将促进未来在揭示感染系统和肿瘤微环境中宿主-微生物相互作用的复杂相互作用方面的发现。该研究结果于2024年发表在ANGEWANDTE CHEMIE-INTERNATIONAL EDITION杂志(IF=16.1,2024)。

图1:Meng等人首次发现了与铁死亡相关的宿主细胞异质性,提示铁死亡可能成为抗A.b感染的临床新靶点。
2、肿瘤-微生物组交互作用探索
微生物可在多种癌症类型中检出,包括在特定无菌的器官中,但它们影响人类肿瘤发生或抗肿瘤应答的背景仍不清楚。Ghaddar等人开发了宿主-微生物组相互作用的单细胞分析(SAHMI),这是一种从宿主组织的单细胞测序中恢复和去噪微生物信号的计算管道[2]。Ghaddar等人使用SAHMI来研究两个人类胰腺癌队列中肿瘤-微生物组的相互作用。Ghaddar等人在一部分肿瘤中发现了体细胞相关细菌,而在非恶性组织中几乎没有这些细菌。这些细菌主要与肿瘤细胞配对,并且它们的存在与细胞类型特异性基因表达和通路活性(包括细胞运动和免疫信号传导)相关。
建模结果表明,肿瘤浸润的淋巴细胞与来自感染组织的T细胞非常相似。最后,利用多个独立的数据集,细胞相关细菌的特征可预测临床预后。肿瘤-微生物组交互作用可能调节胰腺癌的肿瘤发生,对临床治疗具有意义(图2)。该研究结果于2022年发表在CANCER CELL杂志(IF=38.58,2022)。

图2:Ghaddar等使用计算流程SAHMI(宿主-微生物组相互作用的单细胞分析)探索胰腺癌的微生物组。SAHMI识别出与关键癌症特征、免疫活性和预后相关的细菌的肿瘤子集。
3、人类肠道微生物群的适应性状态异质性和宿主-噬菌体活性关联
微生物群落(如居住在人类肠道中的微生物群落)高度多样化和复杂,其中许多对健康和疾病有重要影响。这些微生物群落的作用和功能不仅取决于它们的物种组成和多样性,而且还取决于转录水平上细胞内和细胞间的动态状态。因此,为了全面了解复杂的微生物群落及其宿主,我们迫切需要能够获得单微生物分辨RNA测序信息的强大和可扩展的技术。在该研究中,Shen等人报告了一种基于微滴的smRNA-seq(单微生物RNA测序)方法的开发和应用,该方法能够识别人类样本中的大物种多样性,Shen等人将其命名为smRandom-seq2[3]。Shen等人利用smRandom-seq2为4个人类肠道样本中的细菌和噬菌体测序数据设计了一个三模块计算管道。通过该管道,Shen等人建立了人类肠道微生物组的单细胞水平细菌转录图谱,其中包括29742个单一微生物和329个独特物种。普雷沃菌属(Prevotella)和玫瑰菌属(Roseburia)的不同物种之间的不同适应反应状态以及Phascolarctobacterium succinatutens的内在适应策略异质性被发现。此外,Shen等人在人类肠道微生物组中发现了数百种新的宿主噬菌体转录活性关联。
Shen等人的研究结果表明,smRandom-seq2是一种高通量和高分辨率的smRNA-seq技术,能够高度适应真实世界中复杂的微生物群落,并为理解人类微生物群提供了新的视角(图3)。该研究结果于2024年发表在Protein & Cell杂志(IF=13.6,2024)。

图3:smRandom-seq2和数据分析概述。第一部分是smRandomseq2的实验方法和工作流程。第二部分是smRandom-seq2数据的单一微生物数据分析管道。分类注释(MIC-Anno)后,每个细胞(条形码)将被分配一个分类信息,并进一步使用MIC-Bac和MIC-Phage进行聚类、异质性和宿主-噬菌体关联分析。
4、不可培养的微生物真核生物系统基因组学分析
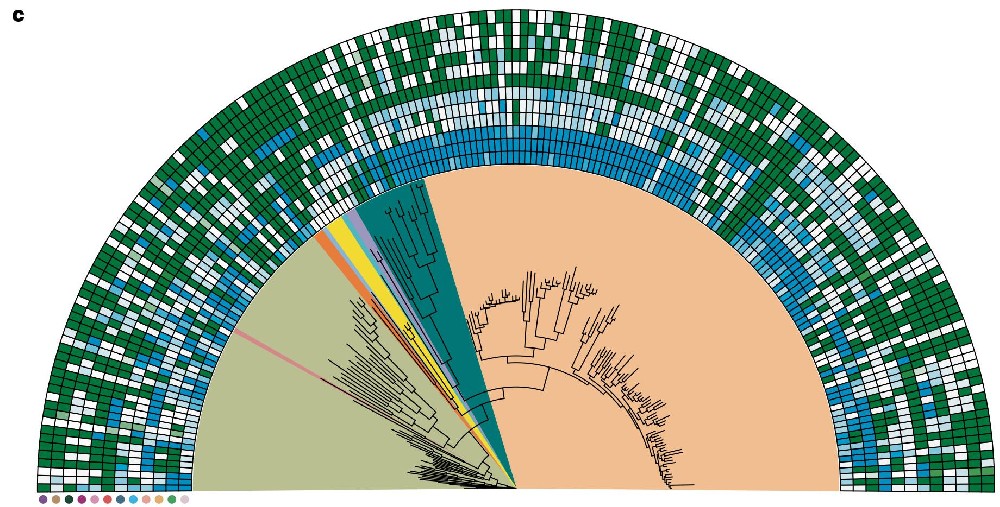
系统发育分析越来越依赖于基因组和转录组数据,以更好地推断微生物真核生物之间的进化关系。然而,这样的系统基因组分析需要稳健的工作流程、生物信息学专业知识和计算能力。鉴于微生物真核生物基因组的复杂性,以及从不可培养谱系的单细胞获得的数据中存在非靶序列(如共生体、猎物),这给微生物真核生物研究带来挑战。
为了解决这些挑战,Shazib等人开发了基于单细胞RNA测序的系统基因组工作流程,整合了从细胞分离到数据管理和物种树推断的所有关键步骤。Shazib等人利用Oligotrichea(一种海洋浮游纤毛虫的多样化群体)的公开可用和新生成的转录组(分别为11和28)评估了他们的工作流程。基于核糖体RNA基因标记,Shazib等人通过转录组序列读码映射重建了这些基因标记,并与Oligotrichea的系统基因组推断进行了比较,这一群体的系统发育关系已经得到了相对充分的研究。
Shazib等人还比较了基于单拷贝蛋白编码基因(精心管理的同源基因)和多拷贝基因(包括同源基因)的系统基因组学分析,分别采用序列拼接和聚结方法(Asteroid)。最后,Shazib等人利用多达1,014个基因家族(GFs)的子集评估了缺失数据在系统基因组学推断中的影响。Shazib等人的所有分析都产生了类似的结果,并且大多数推断出的关系是一致的,并且得到了良好的支持(图4)。该研究结果于2025年发表在MOLECULAR PHYLOGENETICS AND EVOLUTION杂志(IF=3.6,2024)。

图4:从样本采集到基于单细胞RNA测序的系统基因组学分析的步骤。
随着测序技术和微流控技术的不断进步,mscRNA-seq已成为探索微生物异质性、揭示抗药性形成、理解病原感染机制以及工业发酵优化等领域的重要手段。未来,它还将在环境监测、合成生物学、精准医疗(如益生菌开发)等更多领域迎来新的突破。
参考文献:
[1] Meng H, Zhang T, Wang Z, et al. High-Throughput Host-Microbe Single-Cell RNA Sequencing Reveals Ferroptosis-Associated Heterogeneity during Acinetobacter baumannii Infection. Angew Chem Int Ed Engl. 2024;63(18):e202400538. doi:10.1002/anie.202400538
[2] Ghaddar B, Biswas A, Harris C, et al. Tumor microbiome links cellular programs and immunity in pancreatic cancer. Cancer Cell. 2022;40(10):1240-1253.e5. doi:10.1016/j.ccell.2022.09.009
[3] Shen Y, Qian Q, Ding L, et al. High-throughput single-microbe RNA sequencing reveals adaptive state heterogeneity and host-phage activity associations in human gut microbiome. Protein Cell. Published online May 23, 2024. doi:10.1093/procel/pwae027
[4] Shazib SUA, Ahsan R, Leleu M, et al. Phylogenomic workflow for uncultivable microbial eukaryotes using single-cell RNA sequencing - A case study with planktonic ciliates (Ciliophora, Oligotrichea). Mol Phylogenet Evol. 2025;204:108239. doi:10.1016/j.ympev.2024.108239
 技术咨询:
技术咨询: